Chemical Simulations [also known and found as theoretical investigations, model building, molecular modelling, etc] is the single most powerful tool of our times. Simulation via computational tools has rendered the requirement for prototype building and testing obsolete [as well as the requirement of countless chemical experiments in the laboratory]. Current simulation methods offer maximum accuracy and unprecedented speed of calculations. One can predict material composition in order to meet properties requirements within hours of work, in comparison to months of expensive testing in chemical and mechanical laboratories. As an approximation, simulations offers up to 95% cost and time savings during product development, properties prediction, chemical reaction investigations, novel synthetic and manufacturing approaches, and many other phases/ stages.
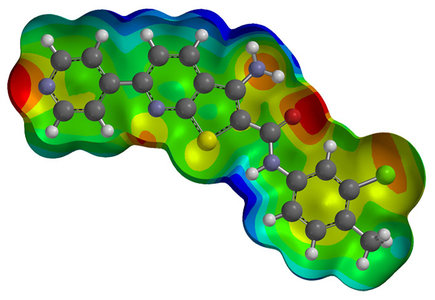
Most important class of chemical simulations are simulations of physical and chemical processes. Simulations of physical and chemical processes as well as prediction of atomic and molecular properties is the foundation of creating new materials, enhancing properties of existing materials, investigating chemical and physical pathways and understanding and explaining nature in general. Chemical Simulations of physical and chemical process is a demanding task. It requires expertise, computational tools, computational chemistry understanding and a significant background in the simulated field. For each calculation, 3D models of all chemical structures are created and optimized using Ab Initio, DFT or semi empirical methods. The two first options provide experimental accuracy, while the third provides quick results of reasonable accuracy. In a second step, the physical or chemical process is simulated using the real 3D models from the previous step and the 'experimental' conditions of interest [pressure, environment, temperature, etc]. A sample of Hydrogen adsorption simulation in novel carbonaceous adsorbents is provided below, from a publication of ours:

Different approaches can be used to perform such simulations and molecular modelling, including Ab Initio, DFT, Molecular Dynamics simulations, and Monte Carlo. Ab Inition and DFT are the best when chemical reactions are expected. Molecular Dynamics simulations is ideal for medium to large size systems, with no chemical reactions and real time investigation of the pathways. Monte Carlo is a stochastic approach that offers speed and accuracy when no chemical reactions are involved. Using simulations and theoretical predictions offers unique advantages to scientists, organizations, academic institutions, and research companies:
- No need for experimental work
- No need for great investments in experimental infrastructure
- Speed in obtaining results
- Constant creation of predictions and databases of results
- Real time monitoring of pathways
- experimental accuracy
- Suggestion and discovery of compounds before experimental discovery
- Quick modification of structures and QSAR work
A specific and very significant category of simulations that we also provide, is known as CFD and it applies to fluid flows as well as other scientific topics. Fluid flows simulation is of the highest importance for chemical, automotive, naval, aerospace, polyemers and plastic industries. Acquiring accurate predictions saves $M for organizations and researchers and reveals optimum constructions and compositions of devices and fluids, respectively. Various CFD computational packages exist, and we have chosen to work with the best of them. Currently we use SimScale for all our CFD simulations.
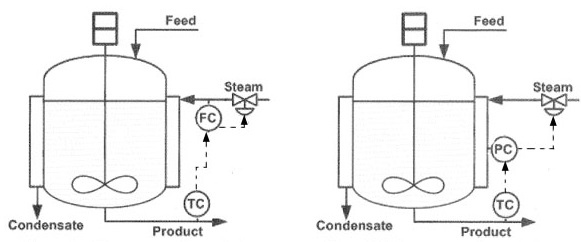
Another category of simulations that is of interest, is the process simulation. Process simulation aims at optimum design of chemical plants by:
- maximizing throughput
- maximizing overall conversion efficiency
- minimizing energy consumption
- reaching optimum device design
- reaching optimum device interconnection
- optimizing heat transfer
- minimizing utilities consumption
- maximizing profit margins
- identifying optimal material properties
- optimizing thermodynamic conversion [exergy]
It has been our choice to work with ASPEN for process simulation and we plan to add more simulation packages in the near future.